The Clinical Approach To The Newborn With Multiple Abnormalities
David Chitayat, MD
The material presented here was first published in the Residents’ Handbook of Neonatology, 3rd edition, and is reproduced here with permission from PMPH USA, Ltd. of New Haven, Connecticut and Cary, North Carolina.
Introduction
Among liveborn infants some 3-5% have major congenital abnormalities and a further 3% have minor abnormalities. In general, 15-20% of the major congenital abnormalities in the liveborn infant involve the central nervous system and the same percentage involves the heart.
With the improvement in the antenatal, intrapartum, postnatal and infant care, the proportion of clinical problems attributable to congenital malformations has increased and about 20% of the paediatric admissions are the result of genetic disorders – single gene disorders, chromosomal abnormalities, exposures to teratogens or multifactorial.
In Canada today, the number of deaths owing to congenital abnormalities approaches the number caused by prematurity. With the continuous improvement in perinatal care, congenital malformations will soon be the main cause of perinatal death.
The presence of multiple congenital abnormalities (MCAs) in a newborn presents difficult diagnostic and management problems. A lack of familiarity with syndromes, in addition to the seemingly endless lists of disorders with confusing eponyms and complicated arrays of features may be overwhelming. This is exacerbated by the highly charged emotional environment in which decisions must be made.
Developing a useful approach to multiple congenital abnormality syndromes can alleviate many of these difficulties by suggesting a list of differential diagnoses, guiding the selection of tests, and providing the most accurate information for parental counselling.
In addition, one must be able to utilize appropriate reference textbooks, computer programs and involve the expertise of a variety of consultants.
The evaluation process of a newborn with MCA should proceed rapidly since decisions regarding intervention and treatment hinge on an accurate diagnosis.
Step I: Obtaining Information
- Obtaining Maternal Medical History
- Obtaining Pregnancy and Reproductive history
- Obtaining Delivery History
- Obtaining Family History
1. Maternal Medical History
A. History of maternal diseases including:
- Type I diabetes mellitus
- Myotonic dystrophy
- PKU (phenylketonuria)
- Autoimmune conditions
B. History of maternal Exposures including:
- Medications
- Alcohol
- Drugs
- Cigarettes
- High temperature
- Infectious diseases
2. Pregnancy history
Pregnancy history can provide information regarding the prenatal onset of congenital abnormalities. This includes:
- Alteration in gestational age: both prematurity and postmaturity.
- Alteration in onset of fetal activity: Fetal activity is usually not felt by the mother until about 18 weeks of gestation (‘quickening’). Fetal activity increases in amount and intensity from that time, reaches a maximum between the 29th and 38th weeks and then decreases somewhat until delivery. Certain structural defects are often associated with delayed onset and\or decreased intensity of fetal activity as well as localization of fetal movement to one particular quadrant of the abdomen. Other examples are defects in brain development or muscle development conditions in which decreased fetal activity is secondary to neuromuscular impairment.
- Abnormalities in the amount of amniotic fluid: oligohydramnios or polyhydramnios.
During the later part of gestation, amniotic fluid is maintained in a constant state of equilibrium by fetal urination, pulmonary efflux and fetal swallowing. Polyhydramnios occurs when the fetus has difficulty swallowing amniotic fluid as in CNS or gastrointestinal anomalies. Oligohydramnios is usually present following chronic leakage of amniotic fluid or abnormalities in the urinary system as in renal agenesis, polycystic kidneys, ureteropelvic or urethral obstruction.
- Findings on detailed fetal ultrasound. Abnormality detected on ultrasound as a result of routine prenatal examinations may be the first indication of significant fetal abnormality. Routine fetal ultrasounds may identify abnormalities and lead to fetal microarray analysis and Whole exome sequencing that may provide important information regarding the pattern and the etiology of the abnormality before birth.
- Findings on prenatal screening – Nuchal translucency measurement, first trimester screening and maternal serum alpha fetoprotein (AFP) screening if doneas well as the levels of the biochemical markers).
3. Delivery History
The delivery history may provide important information regarding the prenatal onset of congenital abnormalities.
A. Presentation:
Breech presentation occurs in 3% of normal-term deliveries. However, it occurs much more frequently in some disorders that adversely affect the form and/or function of the fetus. Structural abnormalities such as hydroce- phalus, would be less compatible with the vertex presentation because of the large head, and joint dislocations (fetal akinesia and arthrogryposis multiplex congenita), which may limit the capacity of the fetus to alter its position. Defects of function include some conditions associated with neuromuscular dysfunction, suchas, trisomy 18, Smith-Lemli-Opitz syndrome, Prader-Willi syndrome, and Zellweger syndrome, among others.
B. Type of delivery:
- Vaginal vs. C-section
- Forceps or vacuum assisted vs. spontaneous
- Induced vs. non-induced
- Other difficulties
C. Prenatal onset of growth deficiency:
There is an increased incidence of prenatal onset malformations as weight for gestational age decreases. In addition, there is a marked increase in mental and neurological defects in small-for-gestational age children who had some structural anomaly.
D. Difficulties in neonatal adaptation:
Children with prenatal onset of structural defects frequently have problems with neonatal respiratory adaptation, which may be secondary to malformations of brain structure. Therefore, one should always be cautious when attributing mental retardation to a perinatal insult in a child who has associated prenatal onset of structural malformations
The physical examination will usually assist in determining if the abnormalities are of prenatal or postnatal onset.
4. Family history
The family history can provide us with the first clue regarding the possible genetic etiology of congenital abnormalities. If either parent has chromosome abnormality or chromosome rearrangement or a known autosomal dominant or the mother has an X-linked disorder, then that particular genetic condition should be anticipated in an offspring. Similarly, a positive family history in other members should raise concerns if the fetus is potentially at risk of the same disorder.
Consanguinity in the parents increases the chance for autosomal recessive conditions as well as multifactorial disorders in their progeny.
Advanced parental age should be perceived as an added risk For women with advanced age there is an increased risk of a chromosome abnormality, such as Down syndrome, and for older fathers there is increased risk of spontaneous mutations in single genes leading to autosomal-dominant syndromes such as achondroplasia and Marfan syndrome.
The pedigree should include
- Names and birthdates, especially of the patients and his first-degree relatives (parents and sibs) and parental sibs
- Age and cause of death of any relative
- Family history of recurrent miscarriages, stillborn, congenital abnormalities and mental retardation
Step 2: Physical and neurological assessment
The most important step in assessing a newborn with MCA is the physical examination:
The clinical assessment of a newborn with multiple congenital abnormalities must be thorough and accurate. Some abnormalities may be quite prominent or involve large areas while others may be rather subtle. The physician should take care to continue to search for the less obvious anomalies even when a major malformation is present. A common pitfall to avoid is the tendency to make the erroneous diagnosis of a well-known syndrome when only one of the major features or several minor features are present.
Particular attention should be paid to the face:
general shape and expression, placement and positioning of facial parts (eye, ear, nose, mouth), skull shape, fontanelle and sutures.
An important part of the physical examination that tends to be ignored is the scalp and upper facial hair pattern.
The origin of each hair follicle, which determines the surface hair directional patterning, is derived from the direction of stretch on the surface skin during the time the hair follicle is growing down from it into the loose underlying mesenchyme at 10 fetal weeks. The slope of each hair follicle and thereby the hair directional patterning is determined by the direction of growth stretch exerted on the surface skin by the development of underlying tissues. For the scalp hair, the patterning relates to the growth in size and form of the underlying brain during the period of 10-16 weeks of fetal growth. By 18 weeks the hair patterning is set. Thus the parietal hair whorl reflects the growth pattern of the brain. In 56% a single parietal hair whorl is located to the left of the midline presumably because the left tend to be slightly larger than the right. In 30% the hair whorl is on the right and in 14% it is in the middle. In 5% there are double hair whorls. There is a correlation between the location of the hair whorl and being right or left handed.
In primary microcephaly the posterior scalp shows a lack of concise whorl, while the anterior scalp showing a marked frontal upsweep.
- The face and neck should be examined in detail including:
- Facial features, symmetry and expression
- Eyebrows, eyes, eyelids, pupil
- Nasal shape, nasal bridge and root, nasal tip, number and shape of nares
- Philtrum
- Lips’ shape, palate (hard and soft), teeth (natal, single incisor), tongue
- Auricles and external auditory meati
- Chin and mandible’s angle and shape
- Torticollis,
- Goiter
- Nuchal hairline
- Webbing
- Nuchal skin
The skin should be carefully examined for texture, pigmentation abnormalities, and other specific lesions. The use of Wood’s lamp is sometimes critical for identifying pigmentation abnormalities that would not otherwise be seen. Examination of the limbs should include careful examination of limbs and digits (number, size, and position and symmetry), creases, dermatoglyphics and nails (size and shape). A general cardiac examination is a necessity. Skeletal examination should include range of motion of joints, pectus deformities, neck length and movement, kyphoscoliosis, and exostosis. When indicated (skeletal dysplasia) the upper/lower ratio and arm span/height should be calculated. In the abdominal examination note hepatosplenomegaly, hernias, abdominal wall defects, or umbilical cord abnormalities. The genitalia should be examined for sexual ambiguity, hypospadias, or cryptorchidism. The back should be examined for scoliosis, kyphosis, pilonidal dimple
Neurological examination including cranial nerves, muscle bulk and tone, DTRs. Neurological findings are often an important indicator of underlying abnormalities. It is often helpful and frequently necessary to examine other family members. Attention is directed towards finding identical features that are present in the child. These may often be quite subtle and present as a “forme frust”. When appropriate, such examination should include first degree relative (parents, siblings) and sometimes second degree relatives (grandparents, aunts, uncles). Photos of relatives are essential for documentation.
Measuring the major features provides objective information on facial characteristics and are important to record. These measurements should be converted to percentiles or standard deviation for accurate comparison among body parts of the patient and among different individuals (see Smith’s Ref. 2).
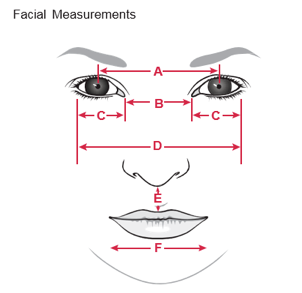
(a) Outer canthal distance
(b) Inner canthal distance
(c) Interpupillary distance
(d) Palpebral fissure length
Step 3: Questions to ask (and answer) after completion of the physical and neurological examination:
A. Is it a Major or Minor abnormality?
The difference between major and minor congenital abnormality is in the severity. A major malformation is a condition of medical, surgical or cosmetic implications. A minor malformation is of less importance and may represent a minor variation.
Some of the abnormalities are not obvious when we take the measurements and it is only when we note the percentile that we find that it is significant. Moreover, sometimes what we think is abnormal may be just normal variation. Facial variations are so frequent that they may be of little significance. On the other hand many minor anomalies may contribute to the diagnosis and can point towards a major congenital abnormality. In one study 14% of newborns were found to have one minor anomaly (reference?), although these babies did not have an increase in the frequency of major congenital abnormalities.
However, only 0.8% of babies had two minor anomalies, and in this subgroup the frequency of a major defect was 10% (5 times more than the control group). It was more striking in the group of babies with three or more minor anomalies. This was found in only 0.5% of babies and 90% of them had one or more major defects.
Thus, the findings of several minor anomalies in the same individual are unusual and often indicate that a more serious problem in morphogenesis has occurred.
Minor external anomalies are most common, in areas of complex and variable features such as the face, auricles, hands, and feet. Before ascribing significance to a given minor anomaly in a patient, it is important to note whether it is found in other family members. Almost any minor defect may occasionally be found as a usual feature in a particular family. This may include clinodactyly, telecanthus, shape and position of the ears, simian crease etc.
B. Is it a single or multiple abnormalities?
It is crucial to differentiate a single anomaly from multiple anomalies, that is, an isolated structural defect versus defects in several areas.
It is usually by the identification of a particular set of abnormalities that the physician can then identify a specific syndrome, which has ramifications for prognosis and recurrence risk. For example, the association of cleft lip with midline defects and polydactyly (as seen in trisomy 13) is a much more serious and complicated situation than the finding of an isolated cleft lip.
Similarly, the findings of a cleft lip associated with lip pits (Van der Wood) has a 50% recurrence risk compared to the approximate 3% recurrence risk of an isolated cleft lip.
Single abnormality: Consider a sporadic or multifactorial condition (i.e. cleft lip, club feet, congenital hip dislocation). Most single primary defects are explained on the basis of multifactorial inheritance, which is thought to carry a recurrence risk for first degree relatives of between 2 and 5%. Thus, recognition that a child’s structural defect represents a single primary defect in development permits the clinician, in the majority of cases, to give recurrence risk figures of between 2-5% to unaffected parents with one affected child.
Before multifactorial risk figures are used for counselling when a single primary defect is recognized, references should be consulted to determine if other risk figures have been reported.
Multiple abnormalities: Consider chromosome abnormality, single gene disorder or exposure to teratogens. In contrast to the anatomical concept of a single primary defect in development, the designation multiple malformation syndrome indicates that the observed structural defects all have the same known or presumed mode of etiology. The defects themselves usually include a number of anatomically unrelated errors in morphogenesis. Recognition that a child has a multiple malformation syndrome is not helpful with respect to recurrence risk counselling unless a specific diagnosis can be made.
Step 4: Describe the abnormality from an etiological point of view
When describing the abnormality from an etiological point of view, it is important that proper terminology be used so that descriptions are accurate and reflect the level of understanding of the pathogenesis.
Malformation: A morphologic defect of an organ, or larger region of the body resulting from an intrinsically abnormal developmental process. The first distinction that must be made in dealing with malformations is whether a given malformation is an isolated finding or part of a more generalized constellation. Most of the common regional malformations are thought to be the result of multifactorial inheritance and include clefting, limb reduction, and neural tube defects. A child with a chromosomal abnormality such as Down syndrome is an excellent example for a generalized malformation, as are many genetic and nongenetic dysmorphology syndromes.
Disruptions: A morphologic defect of an organ or larger region of the body resulting from extrinsic breakdown of or interference with an originally normal developmental process.
Examples of disruptions can be found in the teratogenic syndromes, such as the fetal alcohol syndrome. Mechanical disruption may also be seen with amniotic band syndrome. Most disruptive defects are sporadic events in otherwise normal families, and the recurrence risk is very low. However, the disruption may result in malformations if it occurs early in development, and spontaneous correction is impossible. The prognosis depends on the organ involved.
Deformations: These are abnormal form, shape, or position of body or body part caused by mechanical forces. Deformations are quite common and are frequently self-limiting in nature.
Deformations can be intrinsic or extrinsic in origin. One of the leading causes of deformation of extrinsic origin is an intrauterine constraint as seen in breech presentations and oligohydramnios. However, the oligohydramnios may be the result of fetal abnormality such as renal agenesis, or of extra- fetal etiology such as leakage of amniotic fluid. When the cause is extrafetal, the prognosis is usually good, and 90% of them will be corrected spontaneously.
Sequence: Sequence is a pattern of multiple anomalies derived from a single primary anomaly.
An excellent example here is the Robin sequence, which is the result of micrognathia leading to a small posterior tongue and cleft palate. The primary defect in this case is mandibular hypoplasia, a malformation. Since the tongue is relatively large for the oral cavity, it drops back (glossoptosis), blocking closure of the posterior palatal shelves, resulting in a U-shaped cleft palate.
Association: Nonrandom occurrence of multiple anomalies. For example, the designation of the VATER (vertebral defects, imperforate anus, tracheoesophagel fistula, radial and renal dysplasia) association recognizes that a combination of several or all of these features occurs more often than expected by chance. Another common association is the CHARGE association, an acronym for Coloboma, Heart defect, choanal Atresia, Retardation of growth and development, Genitouri- nary abnormalities, and Ear anomaly/deafness. The importance of associations for the physician is that particular anomalies are found more frequently with other specific anomalies than would be expected by chance. Thus, the identification of any one of these anomalies should prompt a search for the other anomalies with which it is known to be associated. This can often facilitate the evaluation in a given patient and explains the physical features. It should be emphasized that these groupings are associations, not syndromes, and therefore are nonspecific. Indeed, the associations themselves may be found as part of several specific syndromes.
Dysplasia: This refers to the abnormal organization of cell(s) into tissue(s) and its morphological result(s). Examples of the dysplasias are numerous and include neurofibromatosis, connective tissue disorders, and ectodermal dysplasias.
Step 5: Categorizing the Abnormality/ Abnormalities from the Developmental Point of View (Dating the Onset of the abnormality/ies Problems):
A useful approach when confronted with a newborn with MCA is to devise a working hypothesis as to the time of onset of the abnormalities. Thus, it is helpful to differentiate anomalies that occurred prenatally (eg, craniosynostosis) from abnormalities caused at birth (eg, Erb’s palsy, caput succedaneum), and from those that become obvious postnatally (eg, storage diseases).
Stages of normal intrauterine human development
A. The first stage in human development is the pre-implantation period.
This period occurs before and during the first week postconception. This period consists of two stages, gametogenesis and embryogenesis. Gametogenesis is the period of time during which germ cell forms (meiosis) and fertilization occurs, and embryogenesis includes the first cleavage, and morula and blastocyst formation. The blastocyst contains 32-256 cells; however only 25% of these form the inner cell mass which later develops into the embryo.
B. The second stage in human development is the early implantation period which
covers the 2nd and 3rd weeks postconception and includes two major steps:
1) The formation of three germ layers – gastrulation
and
2) the formation of the neural tube – neurolation.
C. The third stage in human development is the embryonic stage. This occurs between
the 4th and 8th weeks of gestation. This stage comprises two main developmental steps:
Morphogenesis: when the head, tail and longitudinal axis are formed
Organogenesis: When the different body organs are being formed.
From the 9th week the conceptus is called a fetus. Prior to this, the conceptus is called an embryo.
Step 6: Categorizing the abnormality/abnormalities from the etiological point of view:
From the etiological point of view multiple malformation syndromes can be divided into five categories (however, the abnormalities in multifactorial conditions should involve one body system):
- Chromosomal
- Single gene
- Multifactorial
- Maternal\teratogens
- Unknown
Recurrence risk depends upon accurate diagnosis, and ranges from low in cases which represent fresh gene mutations or are caused by teratogenes to 100% for the unusual cases of a child with Down syndrome in which the mother is a balanced 21/21 translocation carrier.
A. Chromosomal abnormalities
Certain generalizations are important to consider when thinking about a possible chromosome abnormality:
- Since chromosomes are present in most cells of the body, a chromosome aberration is expected to adversely affect many parts of the body. Consequently, a person with only an in-curved fifth finger and heart defect is very unlikely to have a chromosome abnormality as the etiology of these defects.
- Almost all cases with autosomal chromosome abnormality have lower than normal birth parameters in addition to a near 100% frequency of mental retardation. However, as more experience is gained with chromosomal abnormalities involving very small deletions and duplications this generalization may no longer be valid.
- Most sex-chromosome disorders (XXX, XXY, XYY) have few, if any, initially recognizable defects. Turner syndrome (XO) is an exception.
B. Single Gene Disorder
Mendelian inheritance should be considered when there is a positive family history with an inheritance pattern compatible with single gene transmission, or consanguinity. This may occur within a single generation or through several generations. Transmission between generations and sex predilection should be especially noted.
Autosomal dominant inheritance manifested by a new mutation should be suspected when there is evidence for advanced paternal age.
Autosomal recessive inheritance should be suspected in the presence of consanguinity.
Sex-linked inheritance should be considered when there is no history of affected females and no evidence for male-to-male transmission.
C. Multifactorial inheritance
Many of the common isolated congenital anomalies (e.g. cleft lip, cleft palate, NTD, congenital heart disease) belong to this category. The vast majority of these have been determined empirically from a large collection of family data. However, this mode of inheritance is never responsible for multiple congenital anomalies and can be ruled out when more than one defect is present.
D. Non-traditional patterns of inheritance that do not follow the usual patterns of dominant, recessive, X-linked, or multifactorial include mitochondrial inheritance and genomic imprinting.
E. Maternal diseases
Fetal abnormality can be caused by maternal diseases or medications used to treat maternal diseases. Metabolic maternal diseases causing fetal abnormalities include diabetes mellitus, PKU, cretinism, and galactosemia.
Non metabolic maternal diseases causing fetal abnormalities include maternal myotonic dystrophy, maternal virilizing tumors, and myasthenia gravis.
Medications used by the mother that result in fetal abnormalities include antiepileptic medications, clastogenic medication, accutane and warfarin among others.
F. Teratogens
A teratogenic etiology should be considered whenever the diagnosis is uncertain and whenever a history of prenatal maternal exposure can be elicited. In practice, however, a definitive exposure history to a potential teratogen is usually lacking. Only a relatively small number of teratogens have been directly linked to multiple congenital anomaly syndromes in humans and the best examples are retinoic acid and thalidomide.
In about 40% of the cases with MCA the etiology cannot be found. However, this does not rule out a genetic basis for the findings, and in counselling the family regarding recurrence risk for future pregnancies, this should be pointed out.
A group of multiple congenital anomalies may be diagnosed as a known syndrome in which its genetic cause and\or pattern of inheritance is already known. This emphasizes the importance of utilizing the relevant reference texts and computer programs in which this information may be available
McKusick’s Mendelian Inheritance in Man is a very good resource and is available online at www.ncbi.nlm.nih.gov/omim/ (= OMIM database)
Face2gene: Face2Gene is a suite of phenotyping applications that facilitate comprehensive and precise genetic evaluations. https://www.face2gene.com/
Further work-up that may be suggested according to the findings:
- Photographs are an important source of documentation and are essential in lethal malformations. They are mandatory in increasing the diagnostic yield, can be reviewed by other geneticists or other disciplines if necessary and can help in reducing the mystery surrounding the birth of a malformed child for the parents. The photograph is a crucial adjunct to the written description. In its absence, a review of the medical chart is incomplete and severely limited. Since both normal and abnormal physical features change with age, it is extremely helpful to be able to review these at different ages through photography. Often, family photographs can be obtained for this purpose.
- Radiographic studies: Documentation of skeletal abnormalities should be performed along with additional studies as necessary.
- Laboratory evaluation: Appropriate laboratory studies should be embarked upon, including microarray analysis; to identify a submicrscopic duplication/deletion, including subtelomeric, Whole exome/whole genome sequencing and metabolic studies. Specific metabolic studies are some- times indicated knowing that some facial dysmorphisms are associated with inborn errors of metabolism (ie, Zellweger syndrome, SLO syndrome, X-linked chondrodysplasia punctata [CDPX], etc).
Some practical points in assessing the infant with malformation should be borne in mind:
- Most common diagnosis: We should keep in mind the frequency of different diagnostic categories and avoid jumping into rare diagnostic categories before ruling out the more common causes.
- Decision making is often necessary in the delivery room or nursery concerning the etiology and prognosis of an infant with malformations. The diagnosis should be made as soon as possible based on the clinical assessment since most genetic investigations take time to complete.
- Many malformations produce similar clinical pictures so caution must be used in making a diagnosis.
Following the birth of a child with malformation, the parents and professionals usually share a number of concerns. These include the etiology of the malformation, what can be done, what problems will result from the malformation, the recurrence risk in future pregnancies and the prenatal/preimplantaion diagnosis options. All of these questions demand a thorough and coordinated investigation, consulting the specific specialist, developing skills in dysmorphology, and consulting resource books and computer programs when the diagnosis is not obvious. Most importantly, however, the information should be presented to the parents in a sensitive, supportive, and empathetic manner, keeping in mind potential psychosocial issues arising from the birth of their baby with abnormalities.
Conclusion
According to the World Health Organization (WHO) the term congenital malformations should be confined to structural defects congenital anomalies can be defined as structural or functional anomalies, including metabolic disorders, which are present at the time of birth. Congenital anomalies are an important cause of neonatal mortality. It is a leading cause of fetal loss and contributes significantly to preterm birth, childhood and adult morbidity along with considerable repercussion on the mothers and their families. Parents of children with birth defects face unique challenges and desire to make life better for their kids and express concerns regarding the risk for their future reproductive plans. Some of the challenges parents face involve communication with healthcare professionals, quality of life issues and finding resources and support. Interacting with the parents frequently, informing them that there is nothing that they did or did not do that cause the abnormalities identified and helping them in planning for their future pregnancies by finding the etiology and providing them with prenatal/preimplantation genetic diagnosis helps substantially in lowering anxiety and decision making regarding the approach to their child and their family as a whole.
References:
- Kirpalani, H., Moore, A.M. and Perlman, M., 2007. Residents handbook of neonatology. PMPH-USA
- Diagnostic Dysmorphology by M. Aase
- Smith’s Recognizable Patterns Of Human Malformation by Kenneth Jones
- Practical Genetic Counselling by Peter S. Harper
- A Guide to Genetic Counseling by Diane L. Baker
- Chromosome Abnormalities and Genetic Counseling (Oxford Monographs on Medical Genetics) by R. J. McKinlay Gardner, Grant R. Sutherland
- London dysmorphology database, London neurogenetics database & dysmorphology photo library on CD-ROM
- OMIM database is available online at ncbi.nlm.nih.gov/omim/
- Smith LD, Willig LK, Kingsmore SF Whole-Exome Sequencing and Whole-Genome Sequencing in Critically Ill Neonates Suspected to Have Single-Gene Disorders. Cold Spring Harb Perspect Med. 2015 Dec 18;6(2):a023168. doi: 10.1101/cshperspect.a023168. Review.