Metabolic Disorders
Saadet Mercimek-Andrews, MD
The material presented here was first published in the Residents’ Handbook of Neonatology, 3rd edition, and is reproduced here with permission from PMPH USA, Ltd. of New Haven, Connecticut and Cary, North Carolina.
Various inherited metabolic disorders can present in the neonatal period mimicking hypoxic-ischemic encephalopathy, sepsis, intraventricular hemorrhage, intoxications, or congenital viral infections. Early diagnosis is crucial for two reasons: 1) application of disease specific treatments for treatable inherited metabolic disorders; 2) prognosis and outcomes for non-treatable inherited metabolic disorders. Metabolic investigations should be initiated in newborns, who have not responded to the symptomatic treatment, in the first days of their admission to the neonatal intensive care unit to prevent mortality or long-term morbidity for the treatable inherited metabolic disorders.
When to consider inherited metabolic disorders in newborns
Presence of following clinical and biochemical features:
- Seizures not responding to conventional anti-epileptic drugs
- Unusual body odor such as caramelized sugar (maple syrup urine disease (maple syrup urine disease) or sweaty feet (isovaleric acidemia)
- History of normal delivery and feeding in the first 2-3 days of life, which is followed by progressive encephalopathy, feeding intolerance and vomiting
- Hypoglycemia with normal endocrinological investigations
- Lactic acidosis without history of hypoxia or cardiac or respiratory insufficiency
- Acute hepatic dysfunction including cellular and synthetic liver functions
- Anion gap elevated metabolic acidosis
- Hypoglycemia associated with liver dysfunction and rhabdomyolysis
Family history of:
- Parental consanguinity
- Neonatal or early infantile death with a similar presentation
- Hypoxic-ischemic encephalopathy in the previous child
- History of neonatal deaths in males in the maternal side of the family
Investigations:
Combination of metabolic investigations and type of inherited metabolic disorders are listed below for the differential diagnosis:
Hypoglycemia
Figure 22-1 shows the differential diagnosis of hypoglycemia based on the symptoms. Samples should be collected at the time of the hypoglycemia for metabolic investigations prior to IV glucose to correct hypoglycemia:
- Acylcarnitine profile
- Total and free carnitine
- Free fatty acids
- Beta-hydroxybutyrate
- Ammonium
- Endocrinological investigations for cortisol, growth hormone and insulin
- Immediately after correction of hypoglycemia, urine organic acids and urine ketones should be collected
Metabolic acidosis
Inherited metabolic disorders cause anion gap elevated metabolic acidosis due to accumulation of organic acids, ketone bodies or lactate. Lactate should be >6mmol/L to cause metabolic acidosis. The major category of inherited metabolic disorders presenting with metabolic acidosis:
- Organic acidurias due to accumulation of organic acids: significant hyperammonemia, metabolic acidosis and elevated urine ketones should be present.
- Maple syrup urine disease (MSUD) due to accumulation of branched chain ketoacids: elevated urine ketones should be present in the presence of normal ammonium.
- Ketolysis defects due to accumulation of ketone bodies: mild to moderate hyperammonemia and hypoglycemia should be present.
Hyperammonemia
Inherited metabolic disorders presenting with hyperammonemia:
- Urea cycle disorders with respiratory alkalosis (significant ammonium elevation)
- Organic acidurias with anion gap elevated metabolic acidosis (significant ammonium elevation)
- Ketolysis defects with hypoglycemia and metabolic acidosis (mild to moderate ammonium elevation)
- Fatty acid oxidation defects with hypoglycemia and elevated CK (mild to moderate ammonium elevation)
Lactic acidemia and lactic acidosis
- With normal glucose: multiple carboxylase deficiency, mitochondrial respiratory chain defects, pyruvate dehydrogenase deficiency
- With hypoglycemia: pyruvate carboxylase deficiency, fructose 1,6 biphosphatase deficiency, glycogen storage disease type 1, fatty acid oxidation disorders
Cardiac manifestations
- Arrhythmias: fatty acid oxidation disorders
- Cardiomyopathy:
- Hypertrophic: mitochondrial respiratory chain defects, mitochondrial encephalomyopathies
- Dilated: Fatty acid oxidation disorders, congenital disorders of glycosylation
Hepatic manifestations
- Cholestatic jaundice: Smith-Lemli-Opitz syndrome, congenital disorders of glycosylation, galactosemia, Niemann-Pick disease type C, peroxisomal biogenesis disorders, tyrosinemia type 1, alpha-1-antitripsin,
- Hepatocellular dysfunction or liver failure: galactosemia, tyrosinemia type 1, glycogen storage disease type IV
Table 1: Metabolic investigations, symptoms and inherited metabolic disorders are listed in Table 1.
| Samples | Investigations | Inherited metabolic disorders | Symptoms |
|---|---|---|---|
| Blood | Ammonium | UCD, OAuria | Progressive encephalopathy, vomiting |
| Amino acids | UCD, MSUD, SBD, GE | Progressive encephalopathy, vomiting, IUGR, Seizures, encephalopathy | |
| Homocysteine | IECM, MTHFR def., Homocystinuria | Stroke, seizures | |
| VLCFA | PBD | Dysmorphic features, hepatopathy | |
| CDG test | CDG | Dysmorphic features, hepatopathy | |
| Acylcarnitine profile | OAuria, FAOD | Progressive encephalopathy, vomiting, Hypoglycemia, rhabdomyolysis, hepatopathy, cardiac arrhythmias | |
| Total & free carnitine | OAuria, FAOD, CPT 1 & II def. | Progressive encephalopathy, vomiting, Hypoglycemia, rhabdomyolysis, hepatopathy, CMP | |
| Galactosemia screening | Galactosemia | Hepatopathy, liver insufficiency, jaundice, often E. coli sepsis | |
| Urine | Urine organic acids, | OAuria | Progressive encephalopathy, vomiting |
| Urine ketones | Progressive encephalopathy, vomiting, hypoglycemia | ||
| Sulfocysteine | MoCF def., SOD | Seizures, encephalopathy | |
| Succinylacetone | Tyrosinemia | Hepatopathy, liver insufficiency, jaundice | |
| Alpha-AASA | PDE | Seizures no response to AED | |
| Amino acids | Galactosemia Tyrosinemia |
Hepatopathy, liver insufficiency, jaundice | |
| CSF | Amino acids | GE | Seizures, encephalopathy |
Abbreviations:
AED=anti-epileptic drugs;
CDG=congenital disorders of glycosylation;
CMP=cardiomyopathy;
CPT=carnitine palmitoyl transferase;
def=deficiency;
FAOD= fatty acid oxidation disorders;
GE=glycine encephalopathy;
IECM=inborn errors of cobalamin metabolism;
IUGR=intrauterine growth retardation;
MoCF=molybdenum cofactor;
OAuria=organic aciduria;
PBD=peroxisomal biogenesis disorders;
SBD=serine biosynthesis disorders;
SOD=sulfite oxidase deficiency;
UCD=urea cycle disorders
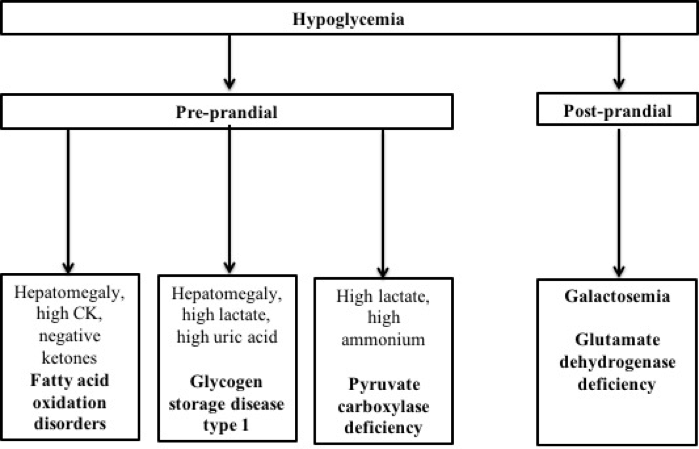
Figure 1 : Differential diagnosis of neonatal hypoglycemia based on the additional clinical and biochemical features suggestive of inherited metabolic disorders.
Additional Readings
- Kirpalani, H., Moore, A.M. and Perlman, M., 2007. Residents handbook of neonatology. PMPH-USA
- Selected topics can be reviewed in GeneReviews at https://www.ncbi.nlm.nih.gov/books/NBK1116/?term=genereviews
- Inborn Metabolic Diseases: Diagnosis and Treatment, 6th Edition, Editors: Jean-Marie Saudubray, Matthias R. Baumgartner, John Walter
- Ogier de Baulny H. Management and emergency treat- ments of neonates with a suspicion of inborn errors of metabolism. Semin Neonatol 2002;7:17–26.
- Prietsch V, Lindner M, Zschocke J, Nyhan WL, Hoffmann GF. Emergency management of inherited metabolic diseases. J Inherit Metab Dis. 2002 Nov;25(7):531-46.
- Jouvet P, Touati G, Lesage F, Dupic L, Tucci M, Saudubray JM, Hubert P. Impact of inborn errors of metabolism on admission and mortality in a pediatric intensive care unit. Eur J Pediatr. 2007 May;166(5):461-5.
- van Rijt WJ, Koolhaas GD, Bekhof J, Heiner Fokkema MR, de Koning TJ, Visser G, Schielen PC, van Spronsen FJ, Derks TG. Inborn Errors of Metabolism That Cause Sudden Infant Death: A Systematic Review with Implications for Population Neonatal Screening Programmes. Neonatology. 2016;109(4):297-302.